For the past four years, I have been unable to post with any regularity. I have dozens of unfinished posts sitting in my drafts folder. I would start with a thought but then get stuck, which had previously been somewhat unusual for me. Now on this first hopeful day I have had for the past four trying years, I am hoping I will be able to post more regularly again.
Prior to what I will now call the Dark Years, I viewed all of history through an economic lens. I bought into the standard twentieth century leftist academic notion that wars, conflicts, social movements, and cultural changes all have economic underpinnings. But I now realize that this is incorrect or at least incomplete. Economics surely plays a role in history but what really motivates people are stories and stories are what led us to the Dark Years and perhaps to get us out.
Trump became president because he had a story. The insurrectionists who stormed the Capitol had a story. It was a bat shit crazy lunatic story but it was still a story. However, the tragic thing about the Trump story (or rather my story of the Trump story) is that it is an unintentional algorithmically generated story. Trump is the first (and probably not last) purely machine learning president (although he may not consciously know that). Everything he did was based on the feedback he got from his Twitter Tweets and Fox News. His objective function was attention and he would do anything to get more attention. Of the many lessons we will take from the Dark Years, one should be how machine learning and artificial intelligence can go so very wrong. Trump’s candidacy and presidency was based on a simple stochastic greedy algorithm for attention. He would Tweet randomly and follow up on the Tweets that got the most attention. However, the problem with a greedy algorithm (and yes that is a technical term that just happens to coincidentally be apropos) is that once you follow a path it is hard to make a correction. I actually believe that if some of Trump’s earliest Tweets from say 2009-2014 had gone another way, he could have been a different president. Unfortunately, one of his early Tweet themes that garnered a lot of attention was on the Obama birther conspiracy. This lit up both racist Twitter and a counter reaction from liberal Twitter, which led him further to the right and ultimately to the presidency. His innate prejudices biased him towards a darker path and he did go completely unhinged after he lost the election but he is unprincipled and immature enough to change course if he had enough incentive to do so.
Unlike standard machine learning for categorizing images or translating languages, the Trump machine learning algorithm changes the data. Every Tweet alters the audience and the reinforcing feedback between Trump’s Tweets and its reaction can manufacture discontent out of nothing. A person could just happen to follow Trump because they like The Apprentice reality show Trump starred in and be having a bad day because they missed the bus or didn’t get a promotion. Then they see a Trump Tweet, follow the link in it and suddenly they find a conspiracy theory that “explains” why they feel disenchanted. They retweet and this repeats. Trump sees what goes viral and Tweets more on the same topic. This positive feedback loop just generated something out of random noise. The conspiracy theorizing then starts it’s own reinforcing feedback loop and before you know it we have a crazed mob bashing down the Capitol doors with impunity.
Ironically Trump, who craved and idolized power, failed to understand the power he actually had and if he had a better algorithm (or just any strategy at all), he would have been reelected in a landslide. Even before he was elected, Trump had already won over the far right and he could have started moving in any direction he wished. He could have moderated on many issues. Even maintaining his absolute ignorance of how govening actually works, he could have had his wall by having it be part of actual infrastructure and immigration bills. He could have directly addressed the COVID-19 pandemic. He would not have lost much of his base and would have easily gained an extra 10 million votes. Maybe, just maybe if liberal Twitter simply ignored the early incendiary Tweets and only responded to the more productive ones, they could have moved him a bit too. Positive reinforcement is how they train animals after all.
Now that Trump has shown how machine learning can win a presidency, it is only a matter of time before someone harnesses it again and more effectively. I just hope that person is not another narcissistic sociopath.
 , where
, where 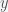 is the phenotype vector,
is the phenotype vector,  is the genotype matrix,
is the genotype matrix,  are all the genetic effects we want to recover, and
are all the genetic effects we want to recover, and  are all the nonadditive components including environmental effects. This is a classic linear regression problem. The problem comes when the number of coefficients
are all the nonadditive components including environmental effects. This is a classic linear regression problem. The problem comes when the number of coefficients  , which consists of some measurements (either a scalar or a vector) at discrete time points
, which consists of some measurements (either a scalar or a vector) at discrete time points 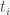 and a proposed model, which produces a time series
and a proposed model, which produces a time series  , where
, where  represents a set of free parameters that changes the function. This model could be a system of differential equations or just some proposed function like a polynomial. The goal is to find the set of parameters that best fits the data and to evaluate how good the model is. Now, the correct way to do this is to use Bayesian inference and model comparison, which can be computed using the MCMC. However, the MCMC can also be used just to get the parameters in the sense of finding the best fit according to some criterion.
represents a set of free parameters that changes the function. This model could be a system of differential equations or just some proposed function like a polynomial. The goal is to find the set of parameters that best fits the data and to evaluate how good the model is. Now, the correct way to do this is to use Bayesian inference and model comparison, which can be computed using the MCMC. However, the MCMC can also be used just to get the parameters in the sense of finding the best fit according to some criterion.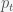 be the price of the fund at time
be the price of the fund at time 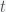 . In dollar cost averaging, you would invest
. In dollar cost averaging, you would invest  dollars at periodic intervals (say each week). So the number of shares you would buy each week is
dollars at periodic intervals (say each week). So the number of shares you would buy each week is 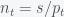 . Over a time period
. Over a time period 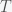 you will have purchased
you will have purchased  shares, which can be rewritten as
shares, which can be rewritten as ![sT E[1/p_t]](https://s0.wp.com/latex.php?latex=sT+E%5B1%2Fp_t%5D&bg=f5f6f7&fg=444444&s=0&c=20201002) , where
, where 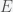 is the expectation value or average. Since you have invested
is the expectation value or average. Since you have invested 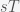 dollars, you have paid
dollars, you have paid ![\tilde p_t=1/E[1/p_t]](https://s0.wp.com/latex.php?latex=%5Ctilde+p_t%3D1%2FE%5B1%2Fp_t%5D&bg=f5f6f7&fg=444444&s=0&c=20201002) per share on average, which is also called the
per share on average, which is also called the 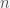 per week then it would cost
per week then it would cost  . Over the time period
. Over the time period  and have
and have  shares. Thus you have paid
shares. Thus you have paid ![\bar{p_t}= E[p_t]](https://s0.wp.com/latex.php?latex=%5Cbar%7Bp_t%7D%3D+E%5Bp_t%5D&bg=f5f6f7&fg=444444&s=0&c=20201002) on average. Now the question is how does the mean
on average. Now the question is how does the mean  compare to the harmonic mean
compare to the harmonic mean  . This is where
. This is where  ,
, ![E[f(x)]\ge f(E[x])](https://s0.wp.com/latex.php?latex=E%5Bf%28x%29%5D%5Cge+f%28E%5Bx%5D%29&bg=f5f6f7&fg=444444&s=0&c=20201002) . A convex function is a function where a straight line joining any two points on the function is greater than or equal to the function. Since
. A convex function is a function where a straight line joining any two points on the function is greater than or equal to the function. Since  is a convex function, this means that
is a convex function, this means that ![E[1/p_t] \ge 1/E[p_t]](https://s0.wp.com/latex.php?latex=E%5B1%2Fp_t%5D+%5Cge+1%2FE%5Bp_t%5D&bg=f5f6f7&fg=444444&s=0&c=20201002) which can be rearrange to show that
which can be rearrange to show that  . So dollar cost averaging is always better than or as good as buying the same number of shares each week. Now of course if you buy on a day when the price is below the harmonic mean of a time horizon you are looking at you will always do better .
. So dollar cost averaging is always better than or as good as buying the same number of shares each week. Now of course if you buy on a day when the price is below the harmonic mean of a time horizon you are looking at you will always do better .
