In one of my very first posts almost a decade ago, I wrote about the end-Permian extinction 250 million years ago, which was the greatest mass extinction thus far. In that post I covered research that had ruled out an asteroid impact and found evidence of global warming, possibly due to volcanos, as a cause. Now, a recent paper in PNAS proposes that a horizontal gene transfer event from bacteria to archaea may have been the main cause for the increase of methane and CO2. This paper is one of the best papers I have read in a long time, combining geological field work, mathematical modeling, biochemistry, metabolism, and evolutionary phylogenetic analysis to make a compelling argument for their hypothesis.
Their case hinges on several pieces of evidence. The first comes from well-dated carbon isotopic records from China. The data shows a steep plunge in the isotopic ratio (i.e ratio between the less abundant but heavier carbon 13 and the lighter more abundant carbon 12) in the inorganic carbonate reservoir with a moderate increase in the organic reservoir. In the earth’s carbon cycle, the organic reservoir comes from the conversion of atmospheric CO2 into carbohydrates via photosynthesis, which prefers carbon 12 to carbon 13. Organic carbon is returned to inorganic form through oxidation by animals eating photosynthetic organisms or by the burning of stored carbon like trees or coal. A steep drop in the isotopic ratio means that there was an extra surge of carbon 12 into the inorganic reservoir. Using a mathematical model, the authors show that in order to explain the steep drop, the inorganic reservoir must have grown superexponentially (faster than exponential). This requires some runaway positive feedback loop that is difficult to explain by geological processes such as volcanic activity, but is something that life is really good at.
The increased methane would have been oxidized to CO2 by other microbes, which would have lowered the oxygen concentration. This would allow for more efficient fermentation and thus more acetate fuel for the archaea to make more methane. The authors showed in another simple mathematical model how this positive feedback loop could lead to superexponential growth. Methane and CO2 are both greenhouse gases and their increase would have caused significant global warming. Anaerobic methane oxidation could also lead to the release of poisonous hydrogen sulfide.
They then considered what microbe could have been responsible. They realized that during the late Permian, a lot of organic material was being deposited in the sediment. The organic reservoir (i.e. fossil fuels, methane hydrates, soil organic matter, peat, etc) was much larger back then than today, as if someone or something used it up at some point. One of the end products of fermentation of this matter would be acetate and that is something archaea like to eat and convert to methane. There are two types of archaea that can do this and one is much more efficient than the other at high acetate concentrations. This increased efficiency was also shown recently to have arisen by a horizontal gene transfer event from a bacterium. A phylogenetic analysis of all known archaea showed that the progenitor of the efficient methanogenic one likely arose 250 million years ago.
The final piece of evidence is that the archaea need nickel to make methane. The authors then looked at the nickel concentrations in their Chinese geological samples and found a sharp increase in nickel immediately before the steep drop in the isotopic ratio. They postulate that the source of the nickel was the massive Siberian volcano eruptions at that time (and previously proposed as the cause of the increased methane and CO2).
This scenario required the unlikely coincidence of several events – lots of excess organic fuel, low oxygen (and sulfate), increased nickel, and a horizontal gene transfer event. If any of these were missing, the Great Dying may not have taken place. However, given that there have been only 5 mass extinctions, although we may be currently inducing the 6th, low probability events may be required for such calamitous events. This paper should also give us some pause about introducing genetically modified organisms into the environment. While most will probably be harmless, you never know when one will be the match that lights the fire.
 ,
,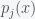 is the probability of amino acid x appearing at position j,
is the probability of amino acid x appearing at position j,  is the probability of amino acids x and y appear at sites j and k, and the sum over x and y is over all the amino acids. Basically, MI is a measure of the “excess” of probability of the occurrence of amino acid x at position j and amino acid y at position k, over what would have occurred if they were statistically independent. One of the problems with mutual information is that you need a lot of data to compute it accurately. Given that we only had a finite number of sequences in each class, error in the MI estimate was expected. So what we did was to set a threshold value for significance compared to the null hypothesis of a set of random sequences.
is the probability of amino acids x and y appear at sites j and k, and the sum over x and y is over all the amino acids. Basically, MI is a measure of the “excess” of probability of the occurrence of amino acid x at position j and amino acid y at position k, over what would have occurred if they were statistically independent. One of the problems with mutual information is that you need a lot of data to compute it accurately. Given that we only had a finite number of sequences in each class, error in the MI estimate was expected. So what we did was to set a threshold value for significance compared to the null hypothesis of a set of random sequences.