Phase transitions may explain why SARS-CoV-2 spreads so fast and why new variants are spreading faster
J.C.Phillips, Marcelo A.Moret, Gilney F.Zebende, Carson C.Chow
Abstract
The novel coronavirus SARS CoV-2 responsible for the COVID-19 pandemic and SARS CoV-1 responsible for the SARS epidemic of 2002-2003 share an ancestor yet evolved to have much different transmissibility and global impact 1. A previously developed thermodynamic model of protein conformations hypothesized that SARS CoV-2 is very close to a new thermodynamic critical point, which makes it highly infectious but also easily displaced by a spike-based vaccine because there is a tradeoff between transmissibility and robustness 2. The model identified a small cluster of four key mutations of SARS CoV-2 that predicts much stronger viral attachment and viral spreading compared to SARS CoV-1. Here we apply the model to the SARS-CoV-2 variants Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Delta (B.1.617.2)3 and predict, using no free parameters, how the new mutations will not diminish the effectiveness of current spike based vaccines and may even further enhance infectiousness by augmenting the binding ability of the virus.
https://www.sciencedirect.com/science/article/pii/S0378437122002576?dgcid=author
This paper is based on the ideas of physicist Jim Phillips, (formerly of Bell Labs, a National Academy member, and a developer of the theory behind Gorilla Glass used in iPhones). It was only due to Jim’s dogged persistence and zeal that I’m even on this paper although the persistence and zeal that ensnared me is the very thing that alienates most everyone else he tries to recruit to his cause.
Jim’s goal is to understand and characterize how a protein will fold and behave dynamically by utilizing an amino acid hydrophobicity (hydropathy) scale developed by Moret and Zebende. People have been developing hydropathy scores for many decades as a way to understand proteins with the idea that hydrophobic amino acids (residues) will tend to be on the inside of proteins while hydrophillic residues will be on the outside where the water is. There are several existing scores but Moret and Zebende, who are physicists and not chemists, took a different tack and found how the solvent-accessible surface area (ASA) scales with the size of a protein fragment with a specific residue in the center. The idea being that the smaller the ASA the more hydrophobic the residue. As protein fragments get larger they will tend to fold back on themselves and thus reduce the ASA. They looked at several thousand protein fragments and computed the average ASA with a given amino acid in the center. When they plotted the ASA vs length of fragment they found a power law and each amino acid had its own exponent. The more negative the exponent the smaller the ASA and thus the more hydrophobic the residue. The (negative) exponent could then be used as a hydropathy score. It differs from other scores in that it is not calculated in isolation based on chemical properties but accounts for the background of the amino acid.
M and Z’s score blew Jim’s mind because power laws are indicative of critical phenomena and phase transitions. Jim computed the coarse-grained hydropathy score (over a window of 35 residues) at each residue of a protein for a number of protein families. When COVID came along he naturally applied it to coronaviruses. He found that the coarse-grained hydropathy score profile of the spike protein of SARS-CoV-1 and SARS-CoV-2 had several deep hydrophobic wells. The well depths were nearly equal with SARS-CoV-2 being more equal than SARS-CoV-1. He then hypothesized that there was a selection advantage for well-depth symmetry and evolutionary pressure had pushed the SARS-CoV-2 spike to be near optimal. He argues that the symmetry allows the protein to coordinate activity better much like the way oscillators synchronize easier if their frequencies are more uniform. He predicted that given this optimality the spike was fragile and thus spike vaccines would be highly effective and that spike mutations could not change the spike much without diminishing function.
My contribution was to write some Julia code to automate this computation and apply it to some SARS-CoV-2 variants. I also scanned window sizes and found that the well depths are most equal close to Jim’s original value of 35. Below is Figure 3 from the paper.

What you see is the coarse-grained hydropathy score of the spike protein which is a little under 1300 residues long. Between residue 400 and 1200 there are 6 hydropathic wells. The well depths are more similar for SARS-CoV-2 and variants than SARS-CoV-1. Omicron does not look much different from the wild type, which makes me think that Omicron’s increased infectiousness is probably due to mutations that affect viral growth and transmission rather than spike binding to ACE2 receptors.
Jim is encouraging (strong arming) me into pushing this further, which I probably will given that there are still so many unanswered questions as to how and why it works, if at all. If anyone is interested in this, please let me know.



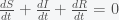 and thus the integral is a constant and given that we started with N people then there will remain N people. This will change if we include births and deaths. Given this conservation law, then the dynamics have three possibilities. The first is that it goes to a fixed point meaning that in the long run the numbers of S, I and R will stabilize to some fixed number and remain there forever. The second is that it oscillates so S, I, and R will go up and down. The final one is that the orbit is chaotic meaning that S, I and R will change unpredictably. For these equations, the answer is the first option. Everything will settle to a fixed point.
and thus the integral is a constant and given that we started with N people then there will remain N people. This will change if we include births and deaths. Given this conservation law, then the dynamics have three possibilities. The first is that it goes to a fixed point meaning that in the long run the numbers of S, I and R will stabilize to some fixed number and remain there forever. The second is that it oscillates so S, I, and R will go up and down. The final one is that the orbit is chaotic meaning that S, I and R will change unpredictably. For these equations, the answer is the first option. Everything will settle to a fixed point.
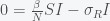

 is large enough then this fixed point is unstable and any amount of I will grow. Assuming I is not zero, we can find another fixed point. Divide I out of the second equation and get
is large enough then this fixed point is unstable and any amount of I will grow. Assuming I is not zero, we can find another fixed point. Divide I out of the second equation and get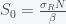

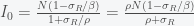

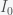 and
and  must be positive implies
must be positive implies 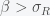 is necessary.
is necessary.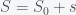 ,
, 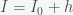 ,
, 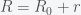 , to get
, to get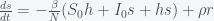

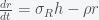
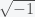 . The only nonlinear term is
. The only nonlinear term is 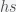 , which we ignore when we linearize. Also by the definition of the fixed point
, which we ignore when we linearize. Also by the definition of the fixed point 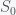 the system then simplifies to
the system then simplifies to 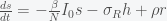

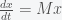 , where
, where  and
and  . The trace of the matrix is
. The trace of the matrix is  so the sum of the eigenvalues is negative but the determinant is zero (since the rows sum to zero), and thus the product of the eigenvalues is zero. With a little calculation you can show that this system has two eigenvalues with negative real part and one zero eigenvalue. Thus, the fixed point is not linearly stable but could still be nonlinearly stable, which it probably is since the nonlinear terms are attracting.
so the sum of the eigenvalues is negative but the determinant is zero (since the rows sum to zero), and thus the product of the eigenvalues is zero. With a little calculation you can show that this system has two eigenvalues with negative real part and one zero eigenvalue. Thus, the fixed point is not linearly stable but could still be nonlinearly stable, which it probably is since the nonlinear terms are attracting.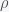 and decreases with
and decreases with 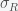 . Thus, as long as the reinfection rate is much smaller than the initial infection rate (which it seems to be), we are headed for a situation where Covid-19 is endemic and will just keep circulating around forever. It may have a seasonal variation like the flu, which is still not well understood and is beyond the simple SIRS equation. If we include death in the equations then there is no longer a nonzero fixed point and the dynamics will just leak slowly towards everyone dying. However, if the death rate is slow enough this will be balanced by births and deaths due to other causes.
. Thus, as long as the reinfection rate is much smaller than the initial infection rate (which it seems to be), we are headed for a situation where Covid-19 is endemic and will just keep circulating around forever. It may have a seasonal variation like the flu, which is still not well understood and is beyond the simple SIRS equation. If we include death in the equations then there is no longer a nonzero fixed point and the dynamics will just leak slowly towards everyone dying. However, if the death rate is slow enough this will be balanced by births and deaths due to other causes. is going to be with us forever, I actually think on the whole the pandemic turned out better than expected, and I mean that in the technical sense. If we were to rerun the pandemic over and over again, I think our universe will end up with fewer deaths than average. That is not to say we haven’t done anything wrong. We’ve botched up many things of course but given that the human default state is incompetence, we botched less than we could have.
is going to be with us forever, I actually think on the whole the pandemic turned out better than expected, and I mean that in the technical sense. If we were to rerun the pandemic over and over again, I think our universe will end up with fewer deaths than average. That is not to say we haven’t done anything wrong. We’ve botched up many things of course but given that the human default state is incompetence, we botched less than we could have. 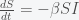

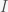 and
and 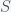 are the infected and susceptible fractions of the initial population, respectively. Behavior greatly affects the rate of infection
are the infected and susceptible fractions of the initial population, respectively. Behavior greatly affects the rate of infection  . If you change
. If you change 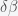 , then the
, then the  and thus the ratio is growing (or decaying) exponentially like
and thus the ratio is growing (or decaying) exponentially like  . The infection rate also appears in the initial reproduction number
. The infection rate also appears in the initial reproduction number  . From a previous
. From a previous 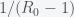 and thus errors in
and thus errors in 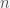 tests on the same subject, the probability of getting more than
tests on the same subject, the probability of getting more than  positive tests if the person is positive is
positive tests if the person is positive is  , where
, where  is the cumulative distribution function of the Binomial distribution (i.e. probability that the number of Binomial distributed events is less than or equal to
is the cumulative distribution function of the Binomial distribution (i.e. probability that the number of Binomial distributed events is less than or equal to  . We thus want to find the minimal
. We thus want to find the minimal  and
and 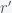 . This means that we need to solve the constrained optimization problem: find the minimal
. This means that we need to solve the constrained optimization problem: find the minimal 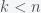 ,
,  and
and 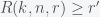 .
. 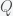 decreases and
decreases and 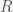 increases with increasing
increases with increasing 

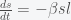
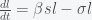
 is the fraction of the population susceptible to SARS-CoV-2 infection and
is the fraction of the population susceptible to SARS-CoV-2 infection and 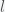 is the fraction of the population actively infectious. Below are simulations of the pandemic progression for R0 = 4 and 2.
is the fraction of the population actively infectious. Below are simulations of the pandemic progression for R0 = 4 and 2.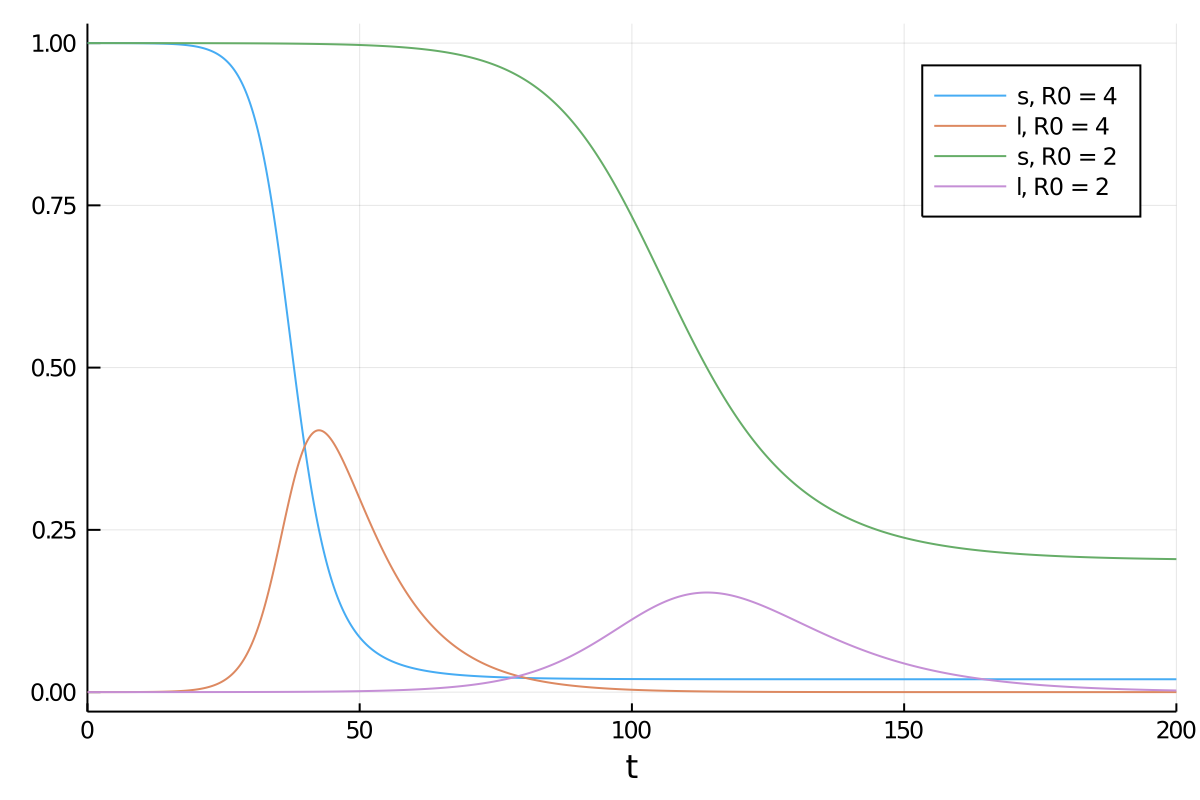
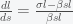 ,
,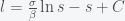 . If we suppose that initially
. If we suppose that initially 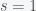 and
and 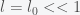 then we get
then we get (*)
(*) is the reproduction number. The total number infected will be
is the reproduction number. The total number infected will be  for
for 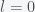 . Rearranging gives
. Rearranging gives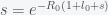
 and ignore
and ignore  we can expand the exponential and solve for
we can expand the exponential and solve for 
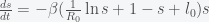
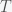 , by separating variables and integrating both sides. The peak is reached when
, by separating variables and integrating both sides. The peak is reached when 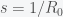 . We must thus compute
. We must thus compute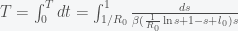
 and
and 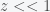 , then we can expand
, then we can expand 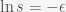 and obtain
and obtain
 . This can be re-expressed as
. This can be re-expressed as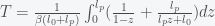
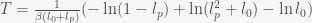
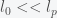 , then we get an expression
, then we get an expression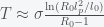
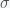 and inversely related to
and inversely related to 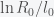 can be big (around 10), which may explain why it takes so long for the pandemic to get started in a region. If the infection rate is very low in a region, the time it takes a for a super-spreader event to make an impact could be much longer than expected (10 times the infection clearance time (which could be two weeks or more)).
can be big (around 10), which may explain why it takes so long for the pandemic to get started in a region. If the infection rate is very low in a region, the time it takes a for a super-spreader event to make an impact could be much longer than expected (10 times the infection clearance time (which could be two weeks or more)).
 into equation (*) and obtain
into equation (*) and obtain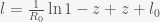
 , which can be expanded to yield
, which can be expanded to yield
 gives the total fraction infected to be
gives the total fraction infected to be
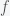 , with a PCR test with 100% sensitivity, which means we do not miss anyone who is positive but we could have false positives. The number of positives we will find is
, with a PCR test with 100% sensitivity, which means we do not miss anyone who is positive but we could have false positives. The number of positives we will find is 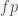 , where
, where  is the prevalence of infectious individuals in a given population. If positive individuals are isolated from the rest of the population until they are no longer infectious with probability
is the prevalence of infectious individuals in a given population. If positive individuals are isolated from the rest of the population until they are no longer infectious with probability  , then the rate of reduction in prevalence is
, then the rate of reduction in prevalence is 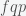 . To reduce the pandemic, this number needs to be higher than the rate of pandemic growth, which is given by
. To reduce the pandemic, this number needs to be higher than the rate of pandemic growth, which is given by  , where
, where  .
. , where
, where  to be somewhere around 0.3 per day. Thus, in the early stages of the pandemic, we would need to test everyone at least two or three times per week, provided positives are isolated. However, if people wear masks and avoid crowds then
to be somewhere around 0.3 per day. Thus, in the early stages of the pandemic, we would need to test everyone at least two or three times per week, provided positives are isolated. However, if people wear masks and avoid crowds then 